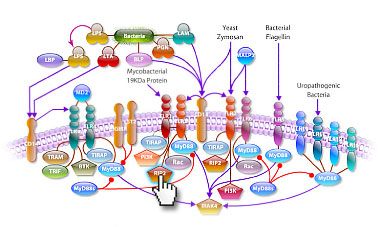
Toll-Like Receptors
TLRs (Toll-like receptors) are transmembrane proteins expressed by cells of the innate immune system, which recognize invading microbes and activate signaling pathways that launch immune and inflammatory responses to destroy the invaders. Toll receptors were first identified in Drosophila. In mammals, the TLR family includes eleven proteins (TLR1-TLR11). Recently, two new members, TLR12 and TLR13 have been discovered in mouse, but not much information is known about them. Mammalian TLRs consist of an extracellular portion containing Leucine-rich repeats, a transmembrane region and a cytoplasmic tail, called the TIR (Toll-IL-1R (Interleukin-1-Receptor)) homology domain. Different TLRs serve as receptors for diverse ligands including Bacterial cell wall components, viral double-stranded RNA and small-molecule antiviral or immune-modulatory compounds (Ref.1). Activation of TLRs occurs after binding of the cognate ligand to the TLR receptors. Upon activation, TLRs activate two major signaling pathways. The core pathway activated by most TLRs lead to the activation of the transcription factor NF-KappaB (Nuclear Factor-KappaB) and the MAPKs (Mitogen-Activated Protein Kinases) p38 and JNK (c-Jun Kinase). The second pathway is activated by TLR3 (Toll-Like Receptor-3) and TLR4 (Toll-Like Receptor-4) and leads to activation of both NF-KappaB and another transcription factor IRF3 (Interferon Regulatory Factor-3), allowing for an additional set of genes to be induced, including antiviral genes such as IFN-Beta (Interferon-Beta). In this way, TLRs can tailor the innate response to pathogens. TLRs that recognize nucleic acids signal from endosomes whereas cell-surface TLRs sense lipids and proteins. Plasma membrane localized TLRs include TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10. Newly discovered TLR11, TLR12 and TLR13 are also believed to be plasma membrane localized, whereas endosomal TLRs include TLR3, TLR7, TLR8 and TLR9 (Ref.2).
Mammalian TLR4 is the signal-transducing receptor activated by the bacterial LPS (Lipopolysaccharide) and LTA (Lipotechoic Acid). Both LPS and LTA first bind to the CD14 (Cluster of Differentiation-14) receptor, which then transfer them to TLR4. TLR4 homo-dimerizes and forms a complex with the protein MD2, and this complex is then transported to the cell surface. LTA can bind directly to CD14, but LPS must be delivered to CD14 by LBP (LPS-Binding Protein). Cells need both MD2 and TLR4 in order to recognize LPS. TLR4 activation engages a set of MyD88 (Myeloid Differentiation Primary-Response Protein-88) adaptor family members, including MyD88, TIRAP (TIR Domain-containing Adapter Protein), TRIF, and TRAM. BTK (Bruton Agammaglobulinemia Tyrosine Kinase) also participate in TLR signaling, although their precise role has yet to be defined (Ref.3). The MyD88 adaptor protein link TLR4 to the IRAK1 (Interleukin-1 Receptor-associated Kinase-1) and IRAK4 Serine/Threonine Kinases, leading to MyD88-dependent pathway. Upon activation of TLR4 by LPS, MyD88 recruits IRAK4, thereby allowing the association of IRAK1 with IRAK4. IRAK4 then induces the phosphorylation of IRAK1. TRAF6 (Tumor Necrosis Factor-Receptor-Associated Factor-6) is also recruited to the receptor complex, which then associate with phosphorylated IRAK1. Phosphorylated IRAK1 and TRAF6 dissociate from the receptor and form a complex with TAK1 (TGF-Beta-Activated Kinase-1), TAB1 (TAK1-Binding Protein-1) and TAB2 (TAK1-Binding Protein-2), which induces the phosphorylation of TAB2 and TAK1. TRAF6, TAK1, TAB1 and TAB2 associate with the Ubiquitin ligases UbC13 (Ubiquitin-Conjugating Enzyme-13) and UEV1A (Ubiquitin-conjugating Enzyme E2-Variant-1). This leads to the ubiquitylation of TRAF6, which induces the activation of TAK1 by interacting with ECSIT (Evolutionarily Conserved Signaling Intermediate in Toll Pathways). TAK1, in turn, phosphorylates both p38 Kinases and JNKs by activating MKK3 (Mitogen-Activated Protein Kinase Kinase-3), MKK6 and MKK7. p38 and JNKs then enter the nucleus and induce the expression of their target genes. TAK1 also phosphorylates the IKK complex (Inhibitor of Kappa Light Polypeptide Gene Enhancer in B-Cells Kinase), which consists of IKK-Alpha (Inhibitor of Kappa Light Polypeptide Gene Enhancer in B-Cells Kinase of Alpha), IKK-Beta (Inhibitor of Kappa Light Polypeptide Gene Enhancer in B-Cells Kinase of Beta) and IKK-Gamma (Inhibitor of Kappa Light Polypeptide Gene Enhancer in B-Cells Kinase of Gamma). The IKK complex then phosphorylates I-KappaB, which leads to its ubiquitylation and subsequent degradation. This causes NF-KappaB translocation to the nucleus where it induces the expression of its target genes. TRIF and TRAM link TLR4 to pathways that lead to TBK1 (TANK Binding Kinase-1) and IRF3 activation (i.e., the MyD88-independent pathway) (Ref.1 & 4).
TLR2 is activated by bacterial LAM (Lipoarabinomannan), BLP (Bacterial Lipoprotein), and PGN (Peptidoglycans). LAM and PGN act on TLR2 through the CD14 receptor, similar to the process followed by the TLR4 with a similar downstream affect. BLP mediates both apoptosis and NF-KappaB activation through TLR2. TLR2 is also responsible for the recognition of the Yeast cell-wall particle Zymosan. Zymosan acts through the CD14 receptor to influence TLR2. TLR2 signals the production of TNF (Tumour Necrosis Factor), through NF-KappaB pathway, from the phagocytized vesicle. TLR6 associates with TLR2 and recognizes diacylated MALP2 (Mycoplasmal macrophage-Activating Lipopeptide-2 kD) along with TLR2. Like TLR4, they also signal through MyD88 and TIRAP. PI3K (Phosphatidylinositde-3 Kinase), RIP2 (Receptor-Interacting Protein-2) and Rac (Ras-Related C3 Botulinum Toxin Substrate) are also involved in TLR6-TLR2 mediated signaling. TLR1 also associates with TLR2 and recognizes the native mycobacterial 19-kDa lipoprotein along with TLR2. TLR1-TLR2 also signals through MyD88, TIRAP, PI3K, RIP2 and Rac. TLR1 and TLR6 may participate in the activation of Macrophages by Gram-positive bacteria. TLR5 is a signaling mediator of bacterial Flagellin, thus activating NF-KappaB and may play a role in resistance to Salmonella infection (Ref.5). Human TLR10 is an orphan member of the TLR family. Genomic studies indicate that TLR10 is in a locus that also contains TLR1 and TLR6, two receptors known to function as coreceptors for TLR2. TLR10 not only homodimerize but can also undergo heterodimerization with TLRs 1 and 2. It has been found to activate gene transcription through MyD88 (Ref.6). TLR9 is responsible for the recognition of CpG islands of bacterial DNA. The extracellular CpG fragment may activate TLR9, thus inducing the endocytosis of the DNA along with TLR9, or perhaps the bacteria is phagocytized and TLR9, which has separately formed on the phagosome, is activated by the CpG islands; whichever the exact method, TLR9 activates the NF-KappaB pathway from the endocytized vesicle. Recently IRF8 (Interferon Regulatory Factor-8) has been shown to be activated by TLR9 through MyD88 (Ref.5).
Besides TLR9, three other TLRs found in endosome are TLR3, TLR7 and TLR8. TLR3 activates immune cells in response to double-stranded Viral RNA. The stimulation of the TLR3 triggers TRIF activation that activates IRF3 through TBK1, independent of MyD88, which lead to the secretion of Ifn-Beta. TRIF also activates RIP1 (Receptor-Interacting Protein-1) and TRAF6, which may further activate NF-KappaB pathway. Small antiviral compounds activate immune cells via the TLR7 MyD88 dependent signaling pathway. TLR7 binds MyD88 and activates IRAF and TRAF6. TRAF6 then activates TANK (also known as I-TRAF). TANK interacts with TBK1 and IKK-Epsilon to activate IRF3. TLR7 or TLR8 may also activate IRF7 through activation of MyD88, BTK and TRAF6, thus inducing antiviral responses by producing Ifn-Alpha (Interferon-Alpha). Recently, Mouse TLR11 has been identified as a participant in defense against Uropathogenic bacteria. The ligands for Mouse TLR12 and TLR13 are currently unknown. Three of them are believed to signal through MyD88 (Ref.7 & 8).
TLR signaling is also negatively regulated by various proteins. The cell-surface receptors ST2 (also known as T1) and SIGIRR (Single ImmunoGlobulin IL-IR-Related molecule (TIR-8)) function as inhibitory receptors, sequestering proteins from signaling complexes and preventing TLR2, TLR4 and TLR9 signaling. IRAKM (Interleukin-1 Receptor-associated Kinase-M), TollIP (Toll-Interacting Protein) and a splice variant of MyD88, known as MyD88s, probably interfere with the recruitment and activation of IRAK4 and IRAK1. Recently, Triad3A, a RING-finger E3 ligase, has been shown to promote ubiquitylation of TLR4 and TLR9, targeting these TLRs for degradation and thereby negatively regulating the intensity and duration of TLR signaling. The balance between activation and inhibition is the key determinant of signal strength of TLR pathways (Ref.9 & 10). In addition to the innate immune response, evidence implicates the involvement of the TLR family in a spectrum of systemic disorders following bacterial infections including Sepsis, Cardiac Ischemia, Peridontitis, and Cerebral palsy. The TLRs that control the onset of an acute inflammatory response are critical antecedents for the development of adaptive acquired immunity. Genetic and developmental variation in the expression of microbial pattern recognition receptors may affect the individual's predisposition to infections in childhood and may contribute to susceptibility to severe neonatal inflammatory diseases, allergies, and autoimmune diseases (Ref.11).
|





Products
Services
Newsletter
