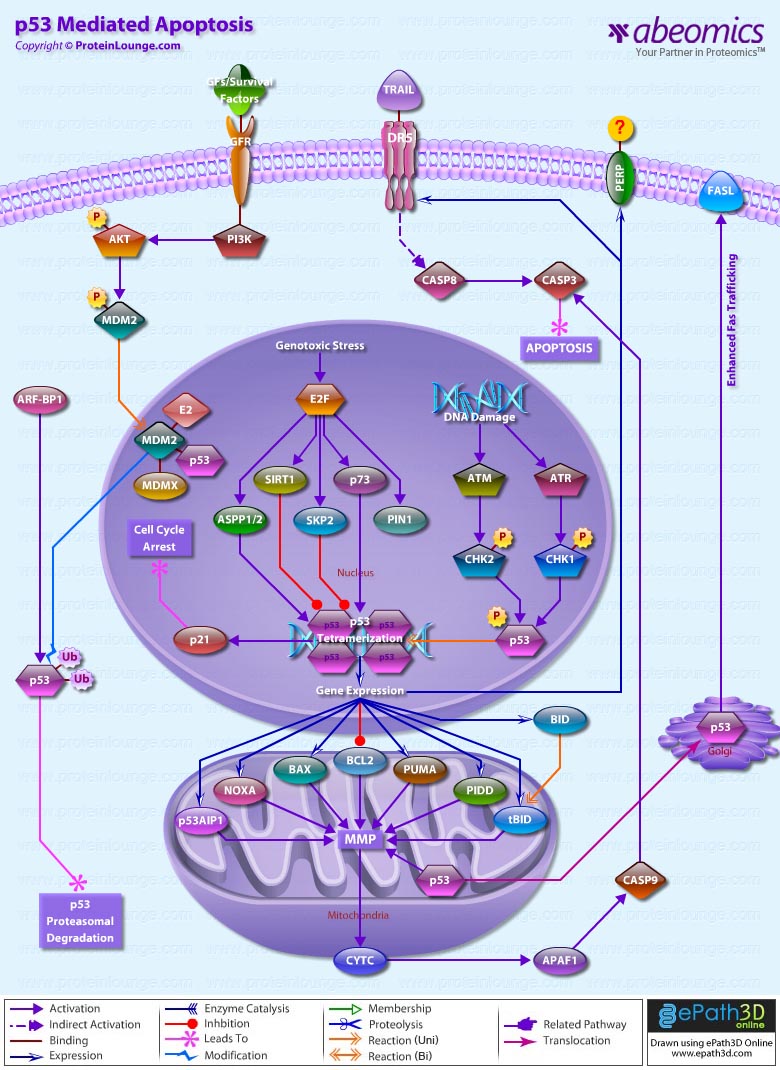
Apoptosis is a genetically controlled mechanism of cell death that is essential for the elimination of unwanted cells during normal development and for the maintenance of tissue homeostasis. One of the major apoptosis signaling pathways involves the p53 tumour suppressor. Tumor protein p53 is a nuclear transcription factor that regulates the expression of a wide variety of genes involved in apoptosis, growth arrest or senescence in response to genotoxic or cellular stress. There are four conserved domains in p53: N-terminal domain, which is required for transcriptional transactivation, a sequence-specific DNA binding domain, a tetramerization domain near the C-terminal end and a C-terminal domain that interacts directly with single stranded DNA. Having a short half-life, p53 is normally maintained at low levels in unstressed mammalian cells by continuous ubiquitylation and subsequent degradation by the 26S Proteasome. Nonphosphorylated p53 is ubiquitylated by the MDM2 (Mouse Double Minute-2) ubiquitin ligase. When the cell is confronted with stress like DNA damage, Hypoxia, Cytokines, metabolic changes, Viral infection, or Oncogenes, however, p53 ubiquitylation is suppressed and p53 is stabilized and accumulates in the nucleus. The N-terminus of p53 may be heavily phosphorylated, whereas the C-terminus may be phosphorylated, acetylated or sumoylated. The ability of p53 to elicit diverse regulatory functions is likely to depend on its phosphorylation pattern, which is conformation dependent. p53 phosphorylation is mediated by several cellular kinases including Chks (Checkpoint Kinases), CSNK1-Delta (Casein Kinase-1-Delta), CSNK2 (Casein Kinase-2), PKA (Protein Kinase A), CDK7 (Cyclin-Dependent Kinase-7), DNA-PK (DNA-Activated- Protein Kinase), HIPK2 (Homeodomain-Interacting Protein Kinase-2) and JNK (Jun NH2-terminal kinase). The main Kinases involved in p53 phosphorylation include Chk1 (Cell Cycle Checkpoint Kinase-1) and Chk2 (Cell Cycle Checkpoint Kinase-2). In response to DNA Damage Chk1 and Chk2, activated by ATR (Ataxia-Telangiectasia and Rad3 Related) and ATM Ataxia Telangiectasia Mutated Gene) respectively, phosphorylates p53. Chk2 can phosphorylate p53 on Ser20, which prevents MDM2 binding and results in p53 stabilization. ATM can also phosphorylate p53 on Ser15, which is required for activation of p53 as a transcription factor and may act synergistically with Ser20 phosphorylation. Once activated p53 binds, as a tetramer, to a p53-binding site. In doing so, it activates expression of downstream genes leading to programmed cell death and/or cell cycle arrest, thus functioning as a tumor suppressor. p53 promotes apoptosis through multiple mechanisms, including transactivation of specific target genes, down-regulation of a distinct set of genes, and transcription-independent mechanisms (Ref. 1 & 2).
p53 stimulates a wide network of signals that act through two major apoptotic pathways: Extrinsic Pathways and Intrinsic Pathways. The Extrinsic pathway involves engagement of particular `death' receptors that belong to the tumor TNFR (Tumor Necrosis Factor Receptor) family and, through the formation of the DISC (Death-Inducing-Signaling-Complex), leads to a cascade of activation of Caspases, including Caspase8 and Caspase3, which in turn induce apoptosis. Most common death receptors involved in extrinsic apoptosis Fas, DR5 (Death Receptor-5) and PERP. The cell-surface receptor Fas (CD95/Apo-1), a member of the TNFR family of receptors, is a key component of the extrinsic death pathway. Fas is activated by binding of its ligand, FasL, which is expressed predominantly by T-Cells. p53 induces Fas mRNA expression by binding to elements found in the promoter and first intron of the Fas gene. This induction occurs in response to Gamma-irradiation, and it appears to be strictly tissue specific. In addition to stimulating Fas transcription, overexpressed p53 may enhance levels of Fas at the cell surface by promoting trafficking of the Fas receptor from the Golgi. This may allow p53 to rapidly sensitize cells to Fas-induced apoptosis before the transcription-dependent effect operates. How p53 promotes Fas trafficking is not understood (Ref. 3 & 4). The second member of this receptor family that is induced by p53 is DR5/Killer, the death-domain-containing receptor for TRAIL (TNF-Related Apoptosis-Inducing Ligand). DR5 is induced by p53 in response to DNA damage and in turn promotes cell death through Caspase8. DR5 induction is cell type specific. Another apoptotic gene, PERP, is induced in response to DNA-damage. PERP is a putative tetraspan transmembrane protein that represents a new member of the PMP-22/Gas family of proteins implicated in cell growth regulation. The kinetics of PERP induction in response to DNA damage and the presence of a p53-responsive element in the PERP promoter support the notion that it is a direct p53 target. The mechanism by which PERP contributes to p53-mediated apoptosis is yet to be defined. Perhaps analogously to Killer and Fas, PERP could serve as a cell death receptor, albeit a different type with four membrane-spanning domains, to receive either autocrine or paracrine signals. Alternatively, a sequence similarity between PERP and the calcium channel suggests that it could have channel or pore activity, perhaps allowing some crucial molecule important for activating apoptosis to pass through. However it exerts its function, PERP might ultimately act by affecting regulators of the apoptotic machinery, such as BAX (Bcl2 Associated-X Protein) or Bcl2, or by directly activating apoptotic effectors like Caspases (Ref. 3, 5 & 6).
The Intrinsic apoptotic pathway is dominated by the Bcl2 family of proteins, which governs the release of CytoC (Cytochrome-C) from the mitochondria. The Bcl2 family comprises anti-apoptotic (pro-survival) and pro-apoptotic members. Family members are classified on the basis of structural similarity to the BH (Bcl2 Homology) domains (BH1, BH2, BH3 and BH4), and a transmembrane domain. The BH3 domain, which is present in all members and is essential for heterodimerization among members, is the minimum domain required for the proapoptotic function. Pro-survival proteins members are most structurally similar to Bcl2, such as BclXL; pro-apoptotic proteins, BAX and BAK (Bcl2 Antagonist Killer-1) are structurally similar to Bcl2 and BclXL and antagonize their pro-survival functions; and the pro-apoptotic `BH3-only' proteins. Intriguingly, a key subset of the Bcl2 family genes are p53 targets, including BAX, Noxa, PUMA (p53-Upregulated Modulator of Apoptosis) and the most recently identified, BID (BH3 Interacting Domain Death Agonist). BAX was the first member of this group shown to be induced by p53, but p53-responsive elements have only recently been unequivocally identified in the BAX gene. In response to stress activation, BAX forms a homodimer and releases CytoC from the mitochondria. CytoC, APAF1 (Apoptotic Protease-Activating Factor-1) and Procaspase9 then form a complex termed the apoptosome, in which Caspase9 is activated and promotes activation of Caspase3, Caspase6 and Caspase7, that after cleavage of vital death substrates induce the final demise of the cell. The requirement for BAX in p53-mediated apoptosis appears to be cell-type dependent (Ref. 7).
The PUMA gene is also directly induced by p53 in response to DNA damage, through p53-responsive elements within the first intron of PUMA. In humans, PUMA encodes two BH3-domain-containing proteins, PUMA-Alpha and PUMA-Beta. BAX is absolutely required for PUMA-mediated apoptosis. PUMA expression promotes mitochondrial translocation and mulitmerization of BAX, culminating in apoptosis induction. BAX thus participates in the death response as an indirect target of p53 through PUMA. Another p53 target gene, Noxa, contains a single p53-responsive element in its promoter and is induced in response to X-ray irradiation. Noxa encodes a BH3-only protein and hence is likely to contribute to p53-mediated apoptosis in a similar manner to PUMA and BAX, although this is yet to be demonstrated. Thus, in response to DNA damage, p53 activates the intrinsic mitochondrial apoptotic pathway by inducing the expression of at least three Bcl2 pro-apoptotic family members, shifting the balance towards pro-apoptotic effects. An important p53 transcription target gene was identified as p53AIP1 (p53-regulated apoptosis-inducing protein 1), which is located in the mitochondrial membrane and is directly involved in p53-dependent mitochondrial apoptosis. Recent evidence has suggested the existence of a transcription-independent pathway of p53-mediated apoptosis. A fraction of the p53 molecules that accumulate in damaged cells translocates to mitochondria, and this translocation is sufficient for p53 to induce permeabilization of the outer mitochondrial membrane through formation of complexes with the protective proteins BclxL and Bcl2, resulting in the release of CytoC into the cytosol. p53 has been shown to transcriptionally repress the antiapoptotic genes Bcl2, BclxL and Survivin (Ref. 8, 9 & 10).
Although Extrinsic and Intrinsic pathways are two separate pathways, recent studies link the Extrinsic and Intrinsic pathways, lending support to the idea of converging rather than distinct pathways. The pro-apoptotic BID connects the activation of the extrinsic death receptor pathway to activation of the mitochondrial-disruption processes associated with the intrinsic pathway. Activation of BID involves cleavage of cytoplasmic BID by Caspase8 to expose a new N-terminal glycine residue, which undergoes post-translational myristoylation. Myristoylated BID translocates to the mitochondria, inserts into the membrane and activates BAX and BAK to initiate mitochondrial events leading to apoptosome formation. The BID gene is transcriptionally regulated by p53 in response to Gamma-irradiation through response elements in the first intron of the human gene. p53 therefore appears to promote the convergence of the intrinsic and extrinsic pathways through BID regulation (Ref.11). Akt also play an important role in regulating p53 pathway under growth/survival conditions and under stress signals. Under normal conditions, activation of Akt can begin with several events, mainly the binding of a Ligand to a Receptor in the cell membrane. Most common Ligands activating Akt include Growth factors, Cytokines, Mitogens and Hormones. Growth factors bind to RTK (Receptor Tyrosine Kinase) and cause autophosphorylation of tyrosine residues on the intracellular domain of the receptor. PI3K (Phosphoinositol 3-Kinase) is recruited to the phosphotyrosine residues (consensus sequence pYXXM) via SH2 domains in the regulatory domain (p85), and is therefore targeted to the inner cell membrane. Binding of the p85 subunit of PI3K to the phosphorylated RTK leads to conformational changes in the catalytic domain of PI3K (p110) and consequent kinase activation. PI3K then phosphorylates membrane bound PIP2 (Phosphatidylinositol-3,4-Bisphosphate) to generate PIP3 (Phosphatidylinositol-3,4,5-Triphosphate). The binding of PIP3 to the PH domain anchors Akt to the plasma membrane and allows its phosphorylation and activation by PDK1 (Phosphoinositide-Dependent Kinase-1). The negative regulation of p53 by AKT is induced in response to survival signals from MDM2. The activation of this pathway leads to the inhibition and destruction of p53. Under stress conditions this pathways is blocked through the cleavage and degradation of Akt, and the inhibition of PI3K through PTEN (Phosphatase and Tensin Homolog). Both of these activities are induced by p53 (Ref. 12 & 13).
Thus, a multitude of mechanisms are employed by p53 to ensure efficient induction of apoptosis in a stage-, tissue- and stress-signal-specific manner. Mutations in p53 are associated with genomic instability and increased susceptibility to cancer. It is the most frequently mutated protein in all cancer with an estimated 60% of all cancers having mutated forms that affect its growth suppressing activities. However some common tumours have a higher incidence such that 90% of cervical and 70% of colorectal are found to have p53 mutations. The p53 protein can be inactivated in several ways, including inherited mutations that result in a higher incidence of certain familial cancers such as Li-Fraumeni syndrome. Certain DNA tumour viruses, such as the human adenovirus and the papilomavirus, bind to and inactivate the protein. Functional p53 is thought to provide a protective effect against tumorigenesis (Ref.14 & 15).
References
1.The role of p53 in apoptosis.
Amaral JD, Xavier JM, Steer CJ, Rodrigues CM.
Discov Med. 2010 Feb;9(45):145-52.
2.p53 Family: Role of Protein Isoforms in Human Cancer.
Wei J, Zaika E, Zaika A.
J Nucleic Acids. 2012;2012:687359. Epub 2011 Oct 9.
3.Placental apoptosis in health and disease.
Sharp AN, Heazell AE, Crocker IP, Mor G.
Am J Reprod Immunol. 2010 Sep;64(3):159-69.
4.p53-mediated delayed NF-?B activity enhances etoposide-induced cell death in medulloblastoma.
Meley D, Spiller DG, White MR, McDowell H, Pizer B, Sée V.
Cell Death Dis. 2010 May 13;1:e41.
5.p53 and the regulation of hepatocyte apoptosis: implications for disease pathogenesis.
Amaral JD, Castro RE, Steer CJ, Rodrigues CM.
Trends Mol Med. 2009 Nov;15(11):531-41. Epub 2009 Oct 12.
6.PERP expression stabilizes active p53 via modulation of p53-MDM2 interaction in uveal melanoma cells.
Davies L, Spiller D, White MR, Grierson I, Paraoan L.
Cell Death Dis. 2011 Mar 31;2:e136.
7.Coordination between cell cycle progression and cell fate decision by the p53 and E2F1 pathways in response to DNA damage.
Zhang XP, Liu F, Wang W.
J Biol Chem. 2010 Oct 8;285(41):31571-80. Epub 2010 Aug 4.
8.PUMA, a potent killer with or without p53.
Yu J, Zhang L.
Oncogene. 2008 Dec;27 Suppl 1:S71-83.
9.Noxa: at the tip of the balance between life and death.
Ploner C, Kofler R, Villunger A.
Oncogene. 2008 Dec;27 Suppl 1:S84-92.
10.p53: the attractive tumor suppressor in the cancer research field.
Ozaki T, Nakagawara A.
J Biomed Biotechnol. 2011;2011:603925. Epub 2010 Dec 6.
11.Post-translational myristoylation: Fat matters in cellular life and death.
Martin DD, Beauchamp E, Berthiaume LG.
Biochimie. 2011 Jan;93(1):18-31. Epub 2010 Nov 5.
12.Oscillations of the p53-Akt network: implications on cell survival and death.
Wee KB, Surana U, Aguda BD.
PLoS One. 2009;4(2):e4407. Epub 2009 Feb 6.
13.Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents.
Degtyarev M, De Mazière A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, Davis DP, Stern HM, Murray LJ, Hoeflich KP, Klumperman J, Friedman LS, Lin K.
J Cell Biol. 2008 Oct 6;183(1):101-16.
14.Mutant p53 gain-of-function in cancer.
Oren M, Rotter V.
Cold Spring Harb Perspect Biol. 2010 Feb;2(2):a001107.
15.Li-fraumeni syndrome.
Malkin D.
Genes Cancer. 2011 Apr;2(4):475-84.