Immune Checkpoint Signaling in Cancer
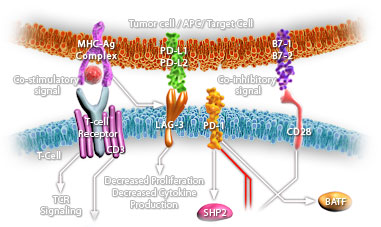
Immune checkpoints are co-stimulatory or inhibitory molecules that serve as regulators of T cells function in the tumor microenvironment. Inhibitory immune checkpoints maintain immune homeostasis, down-regulate anti-tumor responses and prevent autoimmunity (Ref.1). Checkpoint pathways involve costimulatory and inhibitory receptors and their ligands. Costimulatory receptors include CD28 and ICOS (inducible T cell co-stimulator), 4-1BB, OX40, CD27, CD30, CD40, GITR (glucocorticoid inducible TNF receptor-related protein), and HVEM (herpes-virus entry mediator), whereas co-inhibitory receptors include CTLA4, Programmed Death-1 (PD-1), BTLA (B and T lymphocyte attenuator), lymphocyte activation gene-3 (LAG-3), TIM3 (T cell immunoglobulin and mucin domain-containing protein 3), and VISTA (V-domain immunoglobulin suppressor of T cell activation) on T cells (Ref.2). One of the major checkpoints known to augment anti –tumor activity is CTLA4. It is expressed exclusively on T cells. It inhibits B7-CD28–mediated co-stimulatory signals. Signaling through CTLA-4 inhibits IL-2 mRNA production and inhibits cell cycle progression. CTLA-4 can also inactivate T cells via its association with the tyrosine phosphatases SHP1, SHP2, and PP2A. CTLA-4 recruitment of PP2A results in decreased downstream AKT phosphorylation, further dampening the signaling cascade initiated by T cell receptor (TCR) engagement (Ref.3). Another important immune checkpoint molecule that promotes tolerance and dampens T cell immunity at several levels is PD-1. PD-1 is a 55 kDa transmembrane immunoinhibitory protein that plays a major role in tumor immune escape. It is expressed on activated T cells and B cells, NKT cells, tumor-infiltrating lymphocytes, monocytes, and dendritic cells. PD-1 binds to PD-L1 and PD-L2 ligands as well as to the co-stimulatory molecule B7-1 to inhibit T-cell proliferation, survival, and effector functions and to induce apoptosis of tumor-specific T cells. PD-1 recruits the phosphatases SHP1 as well as SHP2, which result in dephosphorylation of the CD3ζ, chain, mediating decreased TCR signaling. PD-1 alters the accumulation of 3-phosphorylated phosphatidylinositol kinase (PI3K). PD-L1 inhibits the PI3K/AKT/ mote signaling pathway (Ref.4). Other checkpoint receptors that are currently under clinical investigation as potential therapeutic targets, either alone or in combination with anti-PD-1 antibodies include TIM3, LAG-3 and VISTA. LAG-3, another immune checkpoint molecule important in the immune response to cancer, is found on activated T cells, B cells, NK cells and plasmacytoid dendritic cells. Its major ligand includes MHC Class II molecules. LSECtin and Galectin-3 may also serve as ligands of LAG-3. A fourth immune checkpoint molecule which is a member of the CD28 family and is a co inhibitory ligand on APCs that suppress T cell responses and induce FOXP3 expression is VISTA. It is expressed mainly in the haematopoietic compartment. VISTA receptors are still unidentified. Anti-VISTA therapy accentuates the development of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. TIM-3, another checkpoint molecule, is a glycoprotein that has both immunoglobulin and mucin domains on its extracellular portion. TIM-3 and LAG-3 have similarly subtle effects in controlling T cell function (Ref.5 & 6). Immune checkpoint signaling attenuates T-cell activation at the molecular, cellular, and physiologic levels to avoid autoimmunity and the destructive effects of an excessive inflammatory response. Immune checkpoint blockade removes inhibitory signals of T-cell activation, allowing them to be activated and recover their antitumor activity. Checkpoint blockade therapy has emerged as a promising strategy for cancer and a variety of malignancies. Clinical trials testing the combination of PD-L1 or CTLA-4 blockade may hold promise for improving treatment efficacy and response. Combinations of checkpoint inhibitors, cancer vaccines, and cytokines, immunotherapy could increase clinical responses to intractable cancers while decreasing treatment-related toxicity (Ref. 7, 8 & 9).
References:
|





Products
Services
Newsletter
