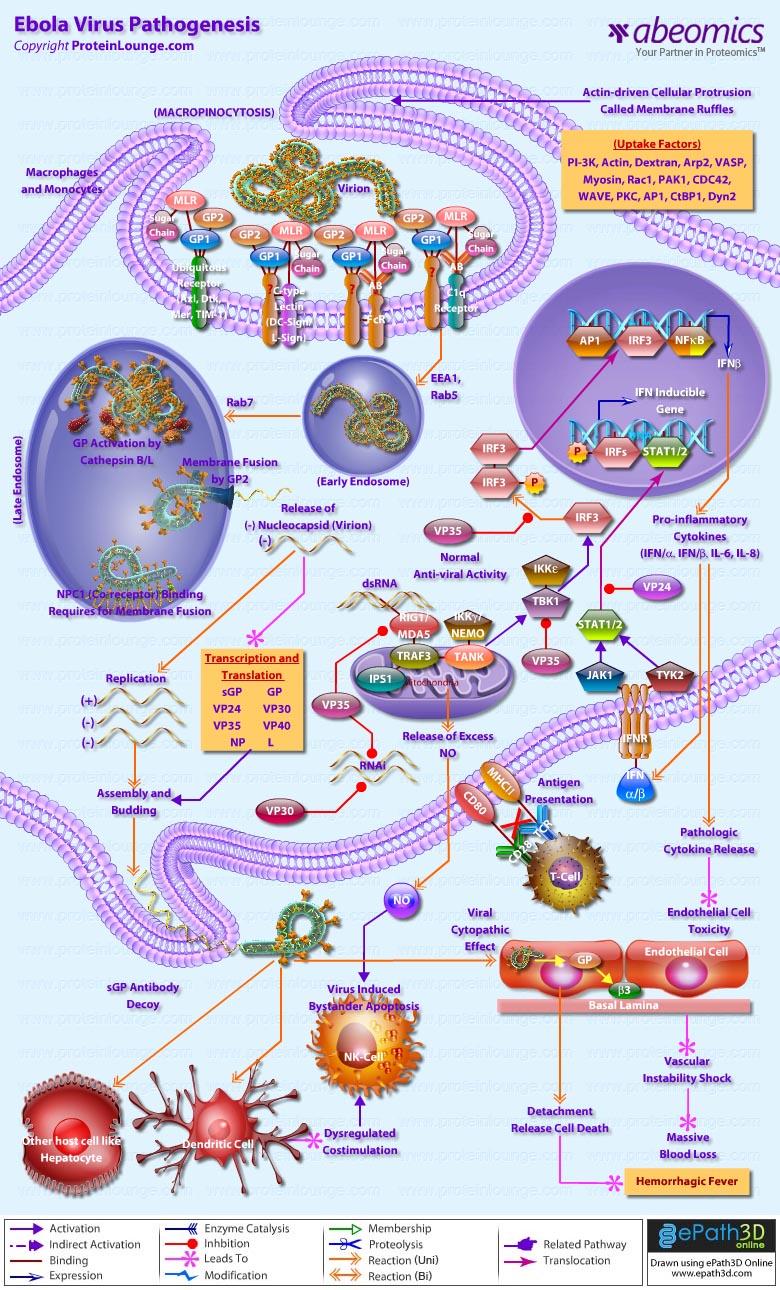
Ebola virus is an aggressive pathogen that causes a highly lethal hemorrhagic fever syndrome in humans and nonhuman primates. Typically, Ebola virus infection runs its course within 14 to 21 days. Infection initially presents with nonspecific flu-like symptoms such as fever, myalgia, and malaise. As the infection progresses, patients exhibit severe bleeding and coagulation abnormalities, including gastrointestinal bleeding, rash, and a range of hematological irregularities, such as lymphopenia and neutrophilia. Cytokines are released when reticulo-endothelial cells encounter virus, which can contribute to exaggerated inflammatory responses that are not protective. Damage to the liver, combined with massive viremia, leads to disseminated intravascular coagulopathy. The virus eventually infects microvascular endothelial cells and compromises vascular integrity. The terminal stages of Ebola virus infection usually include diffuse bleeding, and hypotensive shock accounts for many ebola virus fatalities (Ref.1).
Ebola virus and the related Marburg virus are members of the filovirus family, which are pleomorphic, negative-sense RNA viruses whose genome organization is most similar to the Paramyxoviridae. The Ebola virus genome is 19 kb long, with seven open reading frames encoding structural proteins, including the virion envelope glycoprotein (GP), nucleoprotein (NP), and matrix proteins VP24 and VP40; nonstructural proteins, including VP30 and VP35; and the viral polymerase. The GP open reading frame of Ebola virus gives rise to two gene products, a soluble 60- to 70-kDa dimeric soluble protein (sGP) and a full-length 150- to 170-kDa trimeric protein (GP) that inserts into the viral membrane, through transcriptional editing (Ref.2). This envelope GP spike is expressed at the cell surface, and is incorporated into the virion to drive viral attachment and membrane fusion. It has also been shown as the crucial factor for Ebola virus pathogenicity (Ref. 3). VP40 is located beneath the viral envelope where it helps to maintain structural integrity. It has also been associated with late endosomes and likely mediates filovirus budding due to its ability to induce its own release from cells in the absence of all other viral proteins (Ref.4). The second matrix protein, VP24, has been shown to suppress interferon production. However, interferon interference is not its only function. This protein, along with VP35 and NP are sufficient to form nucleocapsid structures (Ref.5). Lastly, VP24 is necessary for the correct assembly of a functional nucleocapsid. The remaining structural proteins form the nucleocapsid. These are the nucleoprotein NP, the polymerase cofactor VP35, the viral-specific transcription activator VP30 and the viral RNA polymerase L (Ref.2).
As the first step of the viral life cycle, entry into the host cell is a major and multi facet process. Although the mechanism is poorly understood, cellular entry involves uptake by a macropinocytosis-like mechanism. This mechanism is associated with outward extensions of the plasma membrane formed by actin polymerization. These so called membrane ruffles can fold back upon themselves and form a macropinosome upon fusion of the distal loop ends. Virus entry into cells is initiated by the interaction of the viral glycoprotein1 subunit (GP1) with both adherence factors and one or more receptors on the surface of host cells (Ref.6). GP is highly glycosylated with large amounts of N- and O-linked glycans, designated the mucin-like region (MLR). The MLR plays an important role in filovirus entry in to preferred target cells (Ref.7). A variety of C-type lectins (e.g., DC-SIGN, L-SIGN) expressed on key cell types (e.g.,macrophages, DCs) can mediate viral attachment and enhance viral infection (Ref.8). On epithelial cells, TIM-1 serves as a receptor for virus entry (Ref.6). Moreover, members of the Tyro3 receptor tyrosine kinase family (e.g., Axl, Dtk,and Mer) enhances GP-induced macropinocytic uptake (Ref.7).
Internalized virus bearing EBOV GP virions first co-localizes with EEA1 (early endosomal antigen-1)-positive compartments and are then trafficked to Rab5-positive early endosomes. Viral co-localization with perinuclear Rab7/LAMP-1-positive late endosomes can be observed at later times, and delivery to these compartments appears to be important for entry (Ref.8). Within the acidified endosome, the heavily glycosylated GP1 is cleaved to a smaller form by the low pH-dependent cellular proteases Cathepsin B and L, exposing RBS (residues in the receptor binding site), which is a ligand for NPC1 and mediates membrane fusion by the GP2 subunit. NCP1 is crucial for viral membrane fusion and escape from the lysosomal vesicle (Ref.6 & 7). Apart from the conventional micropinocytosis way of entry Filoviruses utilize another mechanism for entry known as antibody-dependent enhancement (ADE) of viral infection, where Filoviruses utilize virus-specific antibodies for their entry into cells in vitro through interaction between anti-GP antibodies and the cellular Fc receptor (FcR) or complement component C1q and its ligand, which likely promotes viral attachment to cells (Ref.7).The energetically unfavorable insertion of the EBOV GP2 fusion loop into host endosomal membranes is followed by the energetically favorable collapse of EBOV GP into a six-helix bundle, allowing for lipid mixing and hemifusion of host and viral membrane lipids. Finally, the hemifused host and viral membranes resolve and a complete pore is formed, through which the viral genomic complex passes into the cytoplasm, allowing the viral replication cycle to continue (Ref.6).
Once the viral and internal cell membranes fuse, the virus particle uncoats and its anti-genome is transcribed into mRNA using nucleocapsid-associated viral proteins. Transcription of the genome is mediated via a complex of VP30, VP35, and the viral polymerase L bound to an NP coated genome. VP30 phosphorylation leads to its dissociation from the VP35/L complex and is the signal to switch from transcription to replication. Following this switch, virus genomes are replicated and coated by NP, VP24, VP30, and VP35 During assembly, L associates with the ribonucleoprotein complex through an interaction with VP35. The ribonucleoproteins, then associate with the matrix protein VP40, and viral particles are extruded through the plasma membrane within lipid raft microdomain regions. Expression and secretion of sGP serves as an antibody decoy for antibodies generated against GP. Viral proteins VP35, VP30, and VP24 are expressed and mediate innate immune avoidance in all cell types. VP35 interferes with RIG-I/MDA-5 signaling and induction of Interferon. Additionally, VP35 and VP30 block the RNAi response against viral gene expression. VP24 acts to inhibit type I and II Interferon (IFN) signaling. This prevents Interferon-induced gene expression and, in antigen-presenting cells, blocks enhancement of antigen presentation to T cells (Ref.9).
During infection, monocytes /macrophages in the lymphoid tissues are early and sustained targets of this deadly virus. The white blood cells respond by releasing large amounts of proinflammatory cytokines that increase permeability of the vascular endothelium, which facilitates easier entry into the virus's secondary targets, endothelial cells. Following GP-mediated macropinocytosis the cell detaches from its neighbors and loses contact with its basement membrane through a mechanism of glycan mediated steric occlusion by GP. The newly created particles then leave via lipid rafts, leaving a destabilized vascular system responsible for the massive blood loss causing hemorrhagic fever characteristic of Ebola patients. The pro-inflammatory cytokines also recruit more macrophages to the area, maximizing the number of cells that ebola can use to spread throughout the body. In the meantime, hepatocytes are being destroyed by the virus, ensuring that these cell signals cannot be cleared from the bloodstream. In addition, infected macrophages release increased amounts of nitric oxide (NO), a gaseous hormone that normally functions in cell communication. However, in high concentrations, NO depresses the mitochondrial membrane potential, causing apoptosis in nearby natural killer cells (Ref. 10 & 11).
References:
1.
Ebola virus pathogenesis: implications for vaccines and therapies.
Sullivan N, Yang ZY, Nabel GJ
J Virol. 2003 Sep;77(18):9733-7. Review. No abstract available.
2.
Ebolavirus: a brief review of novel therapeutic targets.
Kondratowicz AS, Maury WJ.
Future Microbiol. 2012 Jan;7(1):1-4. doi: 10.2217/fmb.11.110. Review.
3.
Studies of ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha.
Yonezawa A, Cavrois M, Greene WC.
J Virol. 2005 Jan;79(2):918-26.
4.
Ebola virus VP40-induced particle formation and association with the lipid bilayer.
Jasenosky LD, Neumann G, Lukashevich I, Kawaoka Y.
J Virol. 2001 Jun;75(11):5205-14.
5.
The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein.
Huang Y, Xu L, Sun Y, Nabel GJ.
Mol Cell. 2002 Aug;10(2):307-16.
6.
Filovirus entry: a novelty in the viral fusion world.
Hunt CL, Lennemann NJ, Maury W.
Viruses. 2012 Feb;4(2):258-75. doi: 10.3390/v4020258. Epub 2012 Feb 7. Review.
7.
Filovirus tropism: cellular molecules for viral entry.
Takada A.
Front Microbiol. 2012 Feb 6;3:34. doi: 10.3389/fmicb.2012.00034. eCollection 2012.
8.
Filovirus entry into cells - new insights.
Miller EH, Chandran K.
Curr Opin Virol. 2012 Apr;2(2):206-14. doi: 10.1016/j.coviro.2012.02.015. Epub 2012 Mar 23. Review.
9.
Camouflage and Misdirection: The Full-On Assault of Ebola Virus Disease.
Misasi J, Sullivan NJ.
Cell. 2014 Oct 23;159(3):477-486. doi: 10.1016/j.cell.2014.10.006. Epub 2014 Oct 16. Review.
10.
Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection.
Geisbert TW, Hensley LE, Larsen T, Young HA, Reed DS, Geisbert JB, Scott DP, Kagan E, Jahrling PB, Davis KJ.
Am J Pathol. 2003 Dec;163(6):2347-70.
11.
Steric shielding of surface epitopes and impaired immune recognition induced by the ebola virus glycoprotein.
Francica JR, Varela-Rohena A, Medvec A, Plesa G, Riley JL, Bates P.
PLoS Pathog. 2010 Sep 9;6(9):e1001098. doi: 10.1371/journal.ppat.1001098.