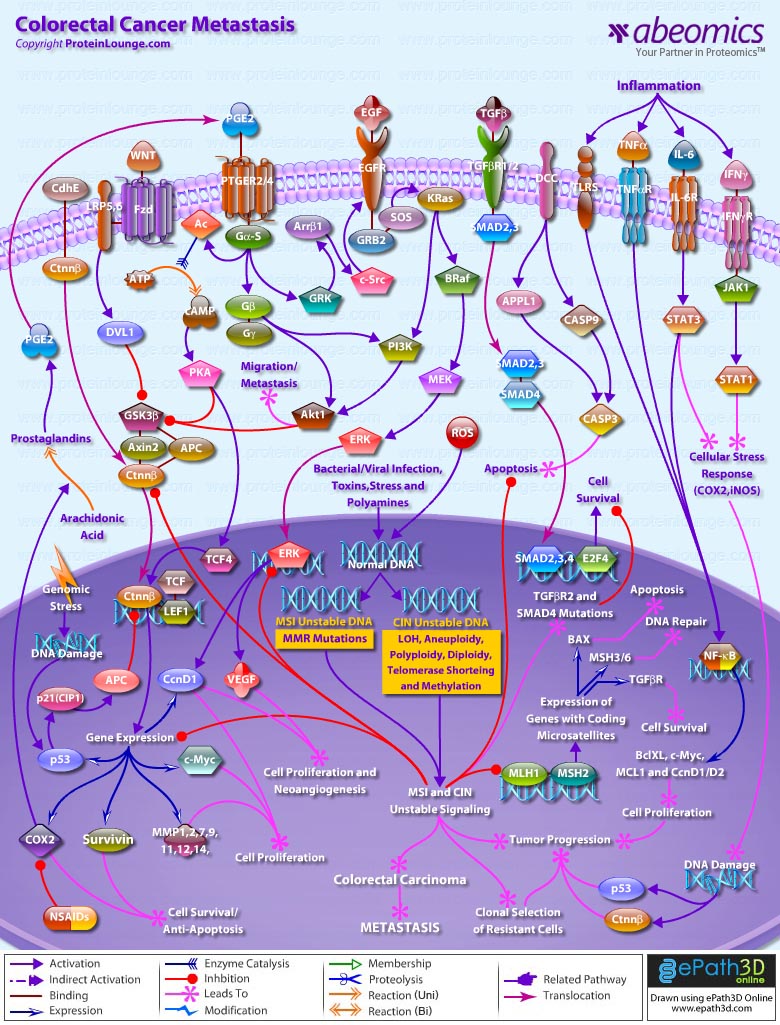
Colorectal cancer represents a relatively well-characterized tumorigenesis paradigm and colorectal carcinoma is one of the leading causes of cancer-related death. Colorectal cancer results from the accumulation of genetic alterations. Genomic instability creates a permissive state in which a potential cancer cell is allowed to acquire enough mutations to become a cancer cell. Several forms of genomic instability are common in colon cancer: MSI (Microsatellite Instability), CIN (Chromosome Instability), and chromosome translocations. MSI occurs in approximately 15% of colon cancers and results from inactivation of the MMR (Mismatch Repair) system secondary to either MMR gene mutations or hypermethylation of MMR gene promoters. It promotes tumorigenesis by generating mutations in target genes that possess coding microsatellite repeats. CIN occurs in most other colon cancers and leads to a different pattern of gene alterations that culminate in tumor formation. It seems to result from mutations in genes that control mitosis, DNA damage repair, centrosome structure and function, and other fundamental processes in DNA replication (Ref.1 & 2). In general the genetic alterations in colorectal cancer progression are determined by one of two separate and distinct underlying pathways of genomic instability. The first pathway, CNI, is characterized by allelic losses, LOH (Loss of Heterozygosity), aneuploidy, diploidy, polyploidy, telomere shortening and methylation. The second pathway, MSI, is characterized by an abundance of subtle DNA mutations. Although the genes causing chromosomal instability remain unknown, MSI is caused by inactivation of a DNA mismatch repair gene (predominantly MLH1 (MutL Homolog-1) or MSH2. Major environmental factors, which influence such gene alternations, include invasion by pathogens (bacterial and viral infections), toxins, stress, polyamines, inflammation, and generation of ROS (Reactive Oxygen Species). The MSI and CIN mutations mainly alter the WNT (Wingless-Type MMTV Integration Site Family), CdhE (Epithelial Cadherin), Prostaglandin, EGFR (Epidermal Growth Factor Receptor), TGF-BetaR (Transforming Growth Factor-Beta Receptor) and DCC (Deleted in Colorectal Carcinoma) signaling routes to facilitate proliferation of colorectal carcinoma. These signaling routes act as check gates in the development of human neoplasia (Ref.3 & 4).
Conceptually, a gatekeeper in human neoplasia refers to a gene that controls the initiation (or is rate-limiting) of a neoplasm in a specific tissue. That is to say, the development of an early neoplasm in this tissue is almost universally associated with (and possibly requires) alteration of this target gene. In contrast, a caretaker refers to a gene that controls the rate of accumulation of genetic alterations (or mutations) during neoplastic progression. Mutation and subsequent inactivation of the APC (Adenomatous Polyposis Coli) tumor suppressor gene in colorectal neoplasia best exemplifies the gatekeeper model. Although APC has a number of putative functions, but the most ascribe role in intestinal neoplasia is to control the WNT signaling pathway via regulation of Ctnn-Beta (Catenin-Beta) levels. In normal cells, the WNT ligands signal via seven transmembrane Fzd (Frizzled) family receptors together with LRP5 (Lipoprotein Receptor-related Protein-5) and LRP6 co-receptors transduce signals through Dsh (Dishevelled) to inhibit GSK3Beta (Glycogen Synthase Kinase-3-Beta) action. In the absence of WNT signaling, Ctnn-Beta is associated with a cytoplasmic complex containing GSK3Beta (Glycogen Synthase Kinase-3-Beta), AXIN (Axis Inhibitor) and the APC (Adenomatous Polyposis Coli) protein, which leads to the degradation of Ctnn-Beta by Proteosome. However, in the presence of WNT signaling, Dsh enhances the phosphorylation of GSK3Beta, which inhibits the ability of GSK3Beta leading to accumulation of free and unphosphorylated Ctnn-Beta in the cytoplasm, which then translocates to the nucleus and associates with TCF and LEF to promote cell survival (anti-apoptosis) and cell proliferation. CdhE also triggers Ctnn-Beta translocation to the nucleus. APC inactivation in tumors by MSI and CIN mutations leads to Ctnn-Beta stabilization, activation of the TCF (Transcription Factor)/LEF (Lymphoid Enhancer-Binding Factor) transcription factor, and subsequent up-regulation of c-Myc, CcnD1 (Cyclin-D1), COX2 (Cyclooxygenase-2), Survivin, p53 and MMP (Matrix Metalloproteinase)-1/2/7/9/11/12/14 (Ref.3,4 & 5). Also genomic stress conditions that induce DNA damage and subsequent p53 activation, leads to the inactivation of p53--->p21(CIP1) (Cyclin Dependent Kinase Inhibitor-p21)--->APC signaling route to up-regulate Ctnn-Beta induced gene transcription and tumor proliferation. Ctnn-Beta, like APC acts as a key mediator in WNT regulation of multiple cellular functions in embryogenesis and tumorigenesis (Ref.6).
Similarly, disruption of COX2 gene (which encodes the key enzyme for conversion of Arachidonic acid to Prostaglandins) and its derived PGE2 (Prostaglandin-E2); this conversion is checked by NSAIDs (Non-Steroidal Anti-Inflammatory Drugs) that inhibit COX2 action), stimulate the growth of cancer cells and promote tumor angiogenesis. PGE2 signals via PTGER2 (Prostaglandin-E Receptor-2) and PTGER4 and activates the Ctnn-Beta/TCF-dependent transcription in colon cancer cells through GN-AlphaS (GN-AlphaS Complex Locus)--->AC (Adenylate Cyclase)--->cAMP (Cyclic Adenosine 3',5'-monophosphate)--->PKA (Protein Kinase-A) pathway. Mechanistically, PGE2 increase the phosphorylation of GSK3Beta and consequently accumulate Ctnn-Beta. In addition, PGE2 induces the expression of TCF4 transcription factor, which forms transcriptionally active complex with Ctnn-Beta (Ref.7). In a parallel cascade the free G-protein Beta and Gamma subunits activate PI3K (Phosphatidylinositde-3 Kinase) and Akt1 (v-Akt Murine Thymoma Viral Oncogene Homolog-1) leading to the phosphorylation of GSK3Beta and the anticipated activation of Ctnn-Beta. Importantly, this pathway seems to be only partially responsible for PGE2-induced Ctnn-Beta-dependent transcriptional activity (Ref.1). Moreover, COX2-derived PGE2 also provides growth advantage to colorectal carcinomas through transactivation of the EGF (Epidermal Growth Factor)/EGFR (Epidermal Growth Factor Receptor) signaling system. When COX2-derived PGE2 binds to and activates the PTGER4, it induces the phosphorylation of the receptor by GRKs (G-Protein-Dependent Receptor Kinases). The phosphorylation on intracellular loops of the PTGER4 attracts Ar-Beta1 (Beta-Arrestin-1). The translocation of Ar-Beta1 to the receptor triggers the dephosphorylation of Ar-Beta1 at Ser14 (Serine-412), thus allowing its association with c-Src. Ar-Beta1 activates c-Src, which then initiates the transactivation of the EGFR and subsequent downstream signaling through Akt1. The EGFR/SOS (Son of Sevenless)/GRB2 (Growth Factor Receptor-Bound Protein-2)--->PI3K--->Akt1 and EGFR/SOS/GRB2--->KRas (Kirsten-Ras)--->PI3K--->Akt1 signaling cascades are involved in the migration and metastasis of colorectal carcinomas (Ref.8). Whereas, activation of the EGFR/SOS/GRB2--->KRas--->BRaf (v-Raf Murine Sarcoma Viral Oncogene Homolog-B1)--->MEK (MAPK/ERK Kinase)--->ERK (Extracellular Signal-Regulated Kinase) signaling cascade is vital for PGE2 induced gene expression of CcnD1 and VEGF (Vascular Endothelial Growth Factor) that plays critical roles in cell proliferation and neoangiogenesis and is vital for establishment of colorectal carcinogenesis as PGE2 and mutated Ctnn-Beta (via MSI/CIN) stimulate the transcription of CcnD1 and VEGF in a synergistic fashion. This indicates that COX2/PGE2 induced EGFR signaling promote the growth of colon cancers through enhancement of tumor angiogenesis (Ref.7).
Apart from the gatekeeper genes, the caretaker genes are also equally important during the manifestation of colon cancers. In this regard, the DNA mismatch repair genes serve as the prototypic model of caretaker genes in human neoplasia (Ref.3). Among the common targets are MLH1 and MSH2 as they control the transcription of genes having coding microsatellite sequences like TGF-BetaR (that promotes cell survival); MSH3/6 (responsible for DNA repair) and BAX (Bcl2 Associated-X Protein) (enhance apoptosis). Signaling by TGF-Beta (Transforming Growth Factor-Beta) superfamily members is disrupted in various intestinal hyperproliferative states (TGF-Beta/TGF-BetaR--->SMAD (Sma and MAD (Mothers Against Decapentaplegic) Related Protein)2/3/4--->E2F4 (E2F Transcription Factor-4)--->cell survival). In the colorectal carcinogenetic sequence, inactivation and mutations of TGF-Beta signaling components occur at the adenoma to carcinoma transition. The most common mutations inactivate TGF-BetaR2, which accounts for the majority of MSI tumors and approximately 55% of MSS (Microsatellite Stable) tumors (Ref.9 & 10). The MSI and CIN mutations are important for clonal selection of resistant cells and tumor progression. Further loss of chromosome 18q21 is well documented in colorectal cancer, and it is suggested that this loss targets the DCC (Deleted in Colorectal Carcinoma), SMAD4 and SMAD2 genes. DCC drives cell death independently of both the mitochondria-dependent pathway and the Death Receptor/Caspase-8 pathway. Moreover, DCC interacts with APPL (Adaptor Protein containing pH domain, PTB domain and Leucine Zipper Motif-1)/DIP13Alpha, Caspase3 and Caspase9 and drives the activation of Caspase3 through Caspase9 without a requirement for CytoC (Cytochrome-C) or APAF1 (Apoptotic Protease Activating Factor-1). DCC acts as a putative tumor suppressor. The importance of SMAD4, a downstream regulator in the TGF-Beta signaling pathway, in colorectal cancer is associated with late-stage or metastatic disease state. SMAD4 mutations occur before colorectal cancers become aneuploid/polyploid, but after the MSI+ and MSI- pathways diverge. MSI+ cancers may diverge first, followed by CIN+ cancers, leaving other cancers to follow a CIN-MSI- pathway (Ref.11, 12 & 13).
Along with MSI and CIN pathways, the inflammatory bowel diseases (IBDs), such as UC (Ulcerative Colitis) and Crohn’s disease, are well known to increase the risk of developing colorectal cancer. Tumor progression is caused by pro-inflammatory cytokines like Ifn-Gamma (Interferon-Gamma), IL-6 (Interleukin-6), TNF-Alpha (Tumor Necrosis Factor-Alpha). Inflammation induced activation of TNF-Alpha/TNF-AlphaR (Tumor Necrosis Factor-Alpha Receptor) and TLRs (Toll-Like Receptors) activate NF-KappaB (Nuclear Factor-KappaB) induced gene activation; whereas, IL-6/IL-6R (Interleukin-6 Receptor) and IFN-Gamma/IFN-GammaR (Interferon-Gamma-Receptor) activate STAT3 (Signal Transducer and Activator of Transcription-3) and STAT1 (through JAK2 (Janus Kinase-2)), respectively. Aberrantly activated STAT3 and NF-KappaB signals participate in oncogenesis through up-regulation of genes such as apoptosis inhibitors (BclXL (Bcl2 Related Protein Long Isoform), MCL1 (Myeloid Cell Leukemia-1)) and cell cycle regulators (c-Myc, CcnD1/D2) to induce cell proliferation and anti-apoptotic factors that leads to tumor progression. Aberrantly activated STAT1 and STAT3 signal induces cellular stress responses (up-regulation COX2, iNOS (Inducible Nitric Oxide Synthase)) and leads to DNA damage (activating p53, Ctnn-Beta), resulting in clonal selection of resistant cells. These signals then orchestrate tumor formation in the colon (Ref.14 & 15). With the many changes that are taking place in dietary habits and lifestyle, colorectal cancer has become a global concern, for which novel treatment needs to be developed in order to add to the therapeutic armamentarium. The development of the RNAi (RNA interference) technology promises new modality of treatment, which may be used in combination with the existing therapies. RNAi is a sequence-specific post-transcriptional gene silencing mechanism, which is triggered by dsRNA (double-stranded RNA) and causes degradation of mRNA homologous in sequence to the dsRNA. This new approach has been successfully adopted to inhibit virus replication and tumorigenicity. DNA vector-based strategies for delivery of siRNA (small interfering RNA) into mammalian cells, further expands the utility of RNAi. siRNAs may be the best tools for target validation in biomedical research and cancer therapeutics, because of their exquisite specificity, efficiency and endurance of gene-specific silencing. Therefore, RNAi provides the opportunity to pursue an exciting new therapeutic approach to treat colorectal cancer (Ref.16).
References:
1. Signal Transduction Cross-talk During Colorectal Tumorigenesis.
Liu X, Lazenby AJ, Siegal GP.
Adv. Anat. Pathol. 2006 Sep;13(5):270-274.
2. Genomic instability and colorectal cancer.
Grady WM, Markowitz S.
Curr. Opin. Gastroenterol. 2000 Jan;16(1):62-67.
3. Carcinogenesis in the GI tract: from morphology to genetics and back again.
Redston M.
Mod. Pathol. 2001 Mar;14(3):236-45.
4. Wnt pathway mutations selected by optimal beta-catenin signaling for tumorigenesis.
Cho KH, Baek S, Sung MH.
FEBS Lett. 2006 Jun 26;580(15):3665-70.
5. Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer.
Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J, Garner RC, Steele RJ, Wolf CR.
Proc. Natl. Acad. Sci. U S A. 2002 Jul 9;99(14):9433-8.
6. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway.
Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, Cordrey A, Zhao Y, Chandraratna RA.
J. Biol. Chem. 2003 Aug 8;278(32):29954-62.
7. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer.
Shao J, Jung C, Liu C, Sheng H.
J. Biol. Chem. 2005 Jul 15;280(28):26565-72.
8. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer.
Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN.
Proc. Natl. Acad. Sci. U S A. 2006 Jan 31;103(5):1492-7.
9. Signaling pathways in intestinal development and cancer.
Sancho E, Batlle E, Clevers H.
Annu. Rev. Cell Dev. Biol. 2004;20:695-723.
10. The role of MLH1, MSH2 and MSH6 in the development of multiple colorectal cancers.
Lawes DA, Pearson T, Sengupta S, Boulos PB.
Br. J. Cancer. 2005 Aug 22;93(4):472-7.
11. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway.
Woodford-Richens KL, Rowan AJ, Gorman P, Halford S, Bicknell DC, Wasan HS, Roylance RR, Bodmer WF, Tomlinson IP.
Proc. Natl. Acad. Sci. U S A. 2001 Aug 14;98(17):9719-23.
12. Mediation of the DCC apoptotic signal by DIP13 alpha.
Liu J, Yao F, Wu R, Morgan M, Thorburn A, Finley RL Jr, Chen YQ.
J. Biol. Chem. 2002 Jul 19;277(29):26281-5.
13. Metastatic colorectal cancer.
Saletti P, Cavalli F.
Cancer Treat. Rev. 2006 Nov;32(7):557-71.
14. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice.
Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, Minoda Y, Sanada T, Yoshioka T, Mimata H, Kato S, Yoshimura A.
J. Exp. Med. 2006 Jun 12;203(6):1391-7.
15. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation.
Itzkowitz SH, Yio X.
Am. J. Physiol. Gastrointest. Liver Physiol. 2004 Jul;287(1):G7-17.
16. RNAi technology: a revolutionary tool for the colorectal cancer therapeutics.
Lv W, Zhang C, Hao J.
World J. Gastroenterol. 2006 Aug 7;12(29):4636-9.
|