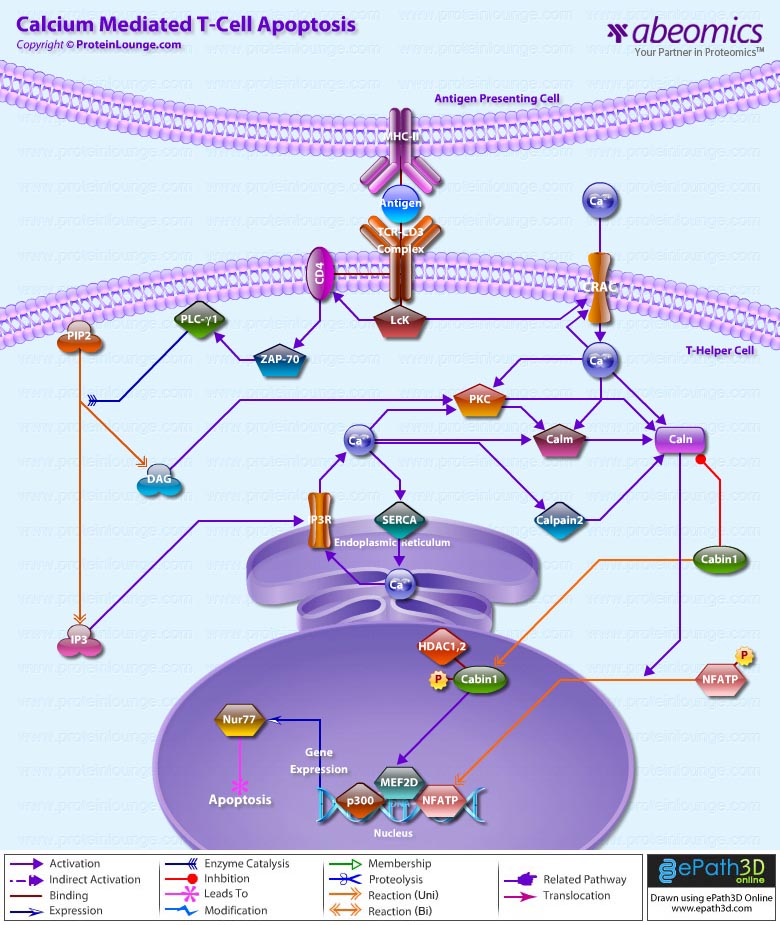
Ca2+ (Calcium) plays a major role in life and death within T-Cells. Elevation of intracellular free Ca2+ is one of the key triggering signals for T-Cell activation by antigen. The binding of antigen, MHC Class-II (Major Histocompatibility Complex Class-II), to the TCR-CD3 (T-Cell Antigen Receptor)-CD3 (CD3 Antigen) triggers the recruitment of a series of tyrosine kinases and substrates to the TCR/CD3/CD4 (CD4 Antigen) Complex, ultimately resulting in the phosphorylation and activation of PLC-Gamma1 (Phospholipase-C-Gamma1). PLC-Gamma1 cleaves PIP2 (Phosphatidylinositol 4,5-bisphosphate) in the plasma membrane to generate DAG (Diacylglycerol), which activates PKC (Protein Kinase-C) and IP3 (Inositol 1,4,5-trisphosphate), that causes entry of Ca2+ to cytosol from two sources: the ER (Endoplasmic Reticulum) and the extracellular space. IP3 generated subsequent to TCR-CD3 stimulation binds to receptors in the ER membrane, opening Ca2+ channels that release Ca2+ to the cytosol (Ref.1). Release is a highly cooperative process, owing to both the binding of multiple IP3 molecules to the tetrameric receptor and to positive feedback by Ca2+ released from the ER. The activity of the IP3R/ITPR (Inositol 1, 4, 5-Triphosphate Receptor) is increased during the early phase of T-Cell activation by Fyn (Fyn Oncogene Related to Src, FGR, YES). IP3R release Ca2+ from intracellular stores and by depleting the stores, trigger prolonged Ca2+ influx through CRAC (Ca2+ Release-Activated Ca2+ Channel/Capacitance-Regulated Activation Channel/Store-Operated Calcium Release-Activated Ca2+/Ca2+ Release-Activated Ca2+ Current) in the plasma membrane. There are distinguished different classes of SOCs (Store-Operated Channels); the class found in T-Cells is the CRAC channel, named for a similar channel originally found in mast cells. It is distinguished from other SOCs primarily by its extremely high Ca2+ selectivity. At short times, Ca2+ signals help to stabilize contacts between T-Cells and Antigen-Presenting Cells through changes in motility and cytoskeletal reorganization (Ref.2).
Mechanisms that remove Ca2+ from the cytosol also exert a powerful influence over the amplitude, duration and dynamics of the net Ca2+ signal. SERCA (Sarco-Endoplasmic Reticulum Ca2+-ATPases) pumps Ca2+ from the cytosol into the ER. SERCA form a significant part of the Ca2+ signaling network in T-Cells by accumulating Ca2+ in the ER, thereby enabling Ca2+ transients via IP3-induced release and controlling store-operated Ca2+ entry. Changes in Ca2+ are associated with a decrease in accumulation of NFATP (Nuclear Factor of Activated T-Cells Pre-existing Component) in the nucleus. NFATP has higher Ca2+ sensitivity but is rapidly reversible. The best current candidates for SOC genes are mammalian homologs of the Drosophila TRP (Transient Receptor Potential) gene. The lack of Ca2+ selectivity or insensitivity to store depletion does not necessarily rule out a role for any particular human TRP in helping to make an endogenous CRAC channel (Ref.3). Focusing on CRAC, which is the only reliably SOC described so far, the crucial experiment will be whether the correct pore properties of CRAC will emerge in a heteromer or a signalplex. This has not yet been shown. There is no doubt that multimerisation occurs for many TRPs, such as TRPC1 (Transient Receptor Potential Cation Channel Subfamily-C Member-1)/4/5, TRPC3/6/7, TRPV1 (Transient Receptor Potential Cation Channel Subfamily-V Member-1)/3, TRPV5/6, TRPM4 (Transient Receptor Potential Cation Channel Subfamily-M Member-4)/5, TRPM6/7 and that heteromer formation changes permeation and kinetic properties of those channels. But so far, no CRAC channel seems to be present in the TRP family since most TRP channels have a low Ca2+ selectivity. In any case, the superficial identification of TRPs as SOCs must be avoided. Then the question remains, what else, if not TRPs? What is the message that links store depletion to channel activation? Although it is too early to say whether CIF (Calcium Influx Factor) directly activates CRAC channels, CIF accelerates CRAC activation relative to activation by EGTA (Ethylene Glycol bis(â-aminoethylether)- N,N,N’,N’-Tetraacetic Acid/ Ethylenebis (Oxyethylenenitrilo) Tetraacetic Acid) alone (Ref.1 & 4).
The “Inside-Out Signaling Pathway” that causes the influx of Ca2+ through specialized CRAC channels provides the persistent Ca2+ signal necessary to maintain NFATP proteins in the nucleus (Ref.2). The major Ca2+ and Caln (Calcineurin)-responsive elements in the Nur77 (Mouse Homolog of Nur77) promoter are binding sites for MEF2D (MADS Box Transcription Enhancer Factor-2 Polypeptide-D). NFATP interacts with MEF2D and enhances its transcriptional activity, offering a plausible mechanism of activation of MEF2D by Caln. NFATP synergizes with MEF2D to recruit the co-activator p300 for the transcription of Nur77. Surprisingly, the enhancement of transcriptional activity of MEF2D by NFATP does not require its DNA-binding activity, proving that NFATP acts as a co-activator for MEF2D. Transient co-expression of p300, MEF2D, NFATP and constitutively active Caln is sufficient to recapitulate TCR signaling for the selective induction of the endogenous Nur77 gene. These results implicate NFATP as an important mediator of T-Cell apoptosis (Ref.5). Binding of Cabin1 (Calcineurin Binding Protein-1) to MEF2D suppresses MEF2D transcriptional activity. However, in the presence of a Ca2+ signal, Calm (Calmodulin) binds to Cabin1, freeing MEF2D to recruit the co-activator p300 for transcriptional activation of MEF2D target genes. Cabin1-MEF2 interaction is required for proper MEF2D induction and phosphorylation after TCR signaling. The COOH-terminal region of Cabin1 interacts with MEF2D and Calm in a mutually exclusive manner. The interaction between Cabin1 and Caln is dependent on both Ca2+ signal and PKC activation, which results in Cabin1 hyperphosphorylation. As Cabin1 is occurs primarily in the nucleus in T-Cells, it interacts only with activated Caln that has translocated into the nucleus (Ref.6).
Capn2 (Calpain-2) cleaves the Caln-binding domain of Cabin1 to activate Caln and elicit Ca2+-triggered cell death. Cabin1 cleavage and Caln activation are suppressed by Capn2 inhibitors. The cleavage of Cabin1 allows Cabin1 to be inactivated and dissociated from the Caln complex, leading to the activation of Caln (Ref.7). In unactivated T-Cells, MEF2D is bound to a transcriptional repression complex consisting of Cabin1, HDAC1 (Histone Deacetylase-1) and HDAC2. Upon TCR signaling and Ca2+ influx, activated Calm binds to Cabin1, releasing it from MEF2D, vacating the MADS/MEF2 domain for association with the coactivator p300. The Ca2+-dependent association and dissociation of two opposing classes of chromatin remodeling enzymes are responsible for tight control of Nur77 transcription, ensuring that thymocytes will not commit to apoptosis in the absence of TCR signaling. Cabin1 also competes with p300 for binding to MEF2D. Activation of MEF2D and the consequent transcription of Nur77 are controlled by the association of MEF2D with the HDACs via the Ca2+-dependent repressor Cabin1 (Ref.8). The role of Ca2+ signaling in regulating MEF2D-dependent gene expression in non-lymphoid tissues still remains the subject of ongoing investigation (Ref.9).
References
1.Calcium signaling mechanisms in T lymphocytes.
Lewis RS.
Annu. Rev. Immunol. 2001;19:497-521.
2.NFAT signaling: choreographing the social lives of cells.
Crabtree GR, Olson EN.
Cell. 2002 Apr;109 Suppl:S67-79.
3.TRP ion channels in the nervous system.
Moran MM, Xu H, Clapham DE.
Curr. Opin. Neurobiol. 2004 Jun;14(3):362-9.
4.Apoptosis.
Wilk S.
Sci. STKE. 2005 May 24;2005(285):tr16.
5.Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis.
Youn HD, Chatila TA, Liu JO.
EMBO J. 2000 Aug 15;19(16):4323-31.
6.Deletion of calcineurin and myocyte enhancer factor 2 (MEF2) binding domain of Cabin1 results in enhanced cytokine gene expression in T cells.
Esau C, Boes M, Youn HD, Tatterson L, Liu JO, Chen J.
J. Exp. Med. 2001 Nov 19;194(10):1449-59.
7.Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death.
Kim MJ, Jo DG, Hong GS, Kim BJ, Lai M, Cho DH, Kim KW, Bandyopadhyay A, Hong YM, Kim do H, Cho C, Liu JO, Snyder SH, Jung YK.
Proc. Natl. Acad. Sci. U S A. 2002 Jul 23;99(15):9870-5.
8.Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2.
Youn HD, Liu JO.
Immunity. 2000 Jul;13(1):85-94.
9.Ca(2+)-dependent gene expression mediated by MEF2 transcription factors.
Blaeser F, Ho N, Prywes R, Chatila TA.
J. Biol. Chem. 2000 Jan 7;275(1):197-209.