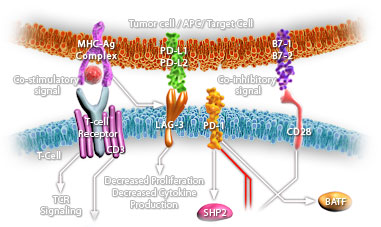
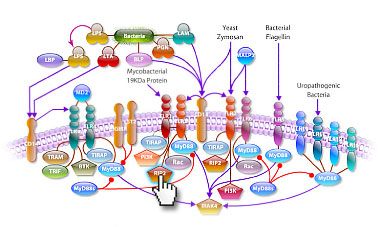
Many proto-oncogenes participate in the regulation of apoptosis and closely intertwined with their actions are various growth factors and other genes that participate in the control of cellular growth. The proto-oncogene c-Myc encodes a transcription factor c-Myc that plays a critical role in multiple cellular processes including cell growth, proliferation, differentiation, and apoptosis. Expression of c-Myc is rapidly induced by mitogens and is down regulated during differentiation. c-Myc activity is sufficient to drive cells into the cell cycle in the absence of growth factors but also induces apoptosis when survival factors are missing. Deregulated expression of c-Myc is associated with a wide array of human cancers. Thr-58 is a major phosphorylation site in c-Myc and is a mutational hotspot in Burkitt's and other aggressive human lymphomas. The quantity of c-Myc is carefully controlled by many mechanisms, and it exerts its oncogenic effects through regulation of genes involved with growth and proliferation (Ref.1).
Expression of c-Myc sensitizes cells to a wide range of mechanistically diverse proapoptotic insults, including DNA damage, death receptor signaling, hypoxia, genotoxic stress, and nutrient deprivation. Two discrete proapoptotic effector pathways mediate this sensitization. One of these involves stabilization of p53 through the ARF (Active Response Factor)/MDM2 (Mouse Double Minute-2) pathway, which serves as a sentinel for genotoxic damage. The second promotes release of Cyto C (Cytochrome-C) from mitochondria into the cytosol, possibly through activation of the pro-apoptotic molecule BAX (BCL2 Associated-X Protein) by a mechanism that is independent of both the Fas-FasL and the DNA damage proapoptotic pathways (Ref.2). Activated BAX within the mitochondrial membrane leads to creation or alteration of membrane pores, resulting in MOMP (Mitochondrial-Outer-Membrane Permeabilization). Once released into the cytosol, Cyto C associates with APAF1 (Apoptotic Protease Activating Factor-1) protein and procaspase-9 to form the apoptosome ('wheel of death'). In the presence of ATP, Caspase 9 is activated, leading to activation of downstream effector caspases, including Caspase 3, which ultimately leads to the degradation of cell components and the demise of the cell. Since the release of Cyto C is the principal target for suppression by survival factors, this pathway acts as a trophic sentinel (Ref.3), triggering Myc-induced apoptosis. c-Myc-induced release of Cyto C is also suppressed by BCL2 (B-Cell CLL/Lymphoma-2)/BCL-XL, which, like survival factors, potently exacerbates c-Myc oncogenicity. Both of these apoptotic pathways share APAF1 and Caspase 9 as final apoptotic effectors downstream of the mitochondrion. Inhibition of this mitochondrial pathway, either by suppression of Cyto C release by survival signals or BCL2/BCL-XL proteins or by incapacity of the downstream mitochondrial apoptotic effector pathway through genetic loss of APAF1 or Caspase 9 (Ref.4) that inhibits c-Myc-induced apoptosis and promotes c-Myc oncogenicity.
Ectopic expression of the death receptor signaling proteins or ligation of the death receptor CD95/Fas triggers the association of the intracellular adaptor protein FADD (FAS-associated death domain), which then recruits procaspase-8, resulting in its auto-activation. Caspase-8 also activates the pro-apoptotic protein BID (BH3 Interacting domain Death agonist), which promotes MOMP. Survival signals that serve to block c-Myc-induced apoptosis include signaling via the IGF1R (Insulin-like Growth Factor-1 Receptor) or activated Ras, which leads to the activation of Akt1 serine/threonine kinase and subsequent phosphorylation of the pro-apoptotic protein BAD (BCL2 Associated Death Promoter). Phosphorylated BAD is sequestered and inactivated by cytosolic 14-3-3 proteins. Anti-apoptotic proteins, BCL2 and BCL-XL also block Cyto C release, possibly through the sequestration of BAX (Ref.5).
The oncogene c-Myc is frequently associated with human malignancies and cancer-related mutations in c-Myc lead to defects in its degradation and thereby contribute to the increase in its cellular level that is associated with the disease. Recent studies suggest that c-Myc is able to activate the cell cycle machinery and intriguingly, its ability to activate glycolysis suggests that in addition to triggering the cell cycle, c-Myc also sustains the fuel necessary to run the cell cycle machinery. Indeed, its ability to enhance the activities of specific enzymes involved in DNA metabolism and other metabolic pathways further suggests that it is a key molecular integrator of cell cycle machinery and cellular metabolism (Ref.6).
References
1.MYC oncogenes and human neoplastic disease.
Nesbit CE, Tersak JM, Prochownik EV.
Oncogene. 1999 May 13; 18(19): 3004-16. Review.
2.c-Myc-induced sensitization to apoptosis is mediated through Cytochrome c release.
Juin P, Hueber AO, Littlewood T, Evan G.
Genes Dev. 1999 Jun 1; 13(11): 1367-81.
3.Proliferation, cell cycle and apoptosis in cancer.
Evan GI, Vousden KH.
Nature. 2001 May 17; 411(6835): 342-8. Review.
4.Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition.
Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW.
Science. 1999 Apr 2; 284(5411): 156-9.
5.c-Myc augments the apoptotic activity of cytosolic death receptor signaling proteins by engaging the mitochondrial apoptotic pathway.
Klefstrom J, Verschuren EW, Evan G.
J Biol Chem. 2002 Nov 8; 277(45): 43224-32. Epub 2002 Aug 28.
6.c-Myc target genes involved in cell growth, apoptosis, and metabolism.
Dang CV.
Mol Cell Biol. 1999 Jan; 19(1): 1-11. Review.